Please
send your answers per email to bioinf@carrot.mcb.uconn.edu,
or hand in a hardcopy
Please let me know, how far you got during the lab.
If most students didn't finish, we may continue this next week!
![]()
a) For a dataset of your choice or here use PhyML (enter "phyml" at the command line) and calculate the tree with the highest likelihood using a model for Among Site Rate Variation (ASRV) that has a proportion of invariant site estimated from the data, and that describes the remaining sites with 4 rate categories that are a discrete approximation of a continuous Gamma distribution whose shape parameter is estimated from the data.
4) Using the same dataset and the same model in TREE-PUZZLE
Invoke TREE-PUZZLE from the command line by typing "puzzle"
Use the tree from (3) as usertree (option k). Take you time in selecting the correct model ! (four rate categories plus invariant sites).
=====================This is how far I expect everyone to get today!==================
4B) Strict Molecular Clock
Repeat the analyses from above, but estimate if a strict molecular clock is compatible with the data (option z).
Select the pinvar and alpha from the previous analysis.
For a large data set, this might
take some time (about 20 minutes for archaea_euk.phy). Start it, once it is running,
open a new ssh connection , and qrsh to a different node (preferred), alternatively
you can send the process (running puzzle) into background. For the latter, stop
the process in foreground by pressing down <ctrl> and <z> simultaneously.
Then restart the process in background by typing
bg
%1
While waiting, continue with 5 below.
5) ML mapping
Use a dataset of your choice or use testseq5.phy.
The latter file contains vacuolar/archaeal ATPases from
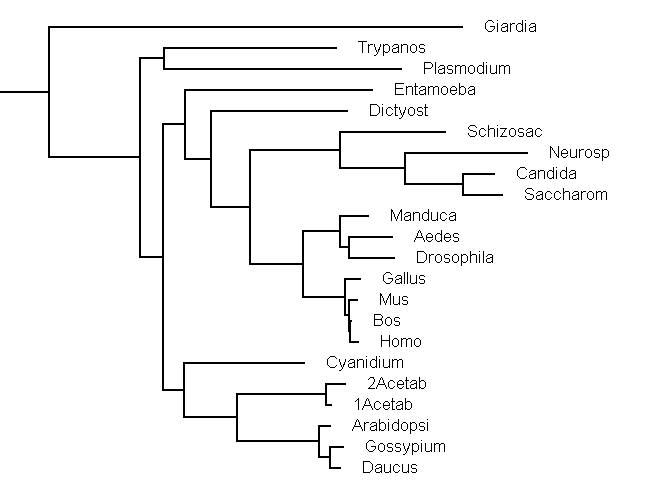
the following pro- and eukaryotes.
Daucus
carota, Arabidopsis
thaliana, Gossypium hirsutum are plants;
Acetabularia acetabulum is a green
and Cyanidium caldarium is a red algae
Mus, Homo, Bos (mammals)
Gallus (bird), Drosophila, Aedes (insects) are animals
Saccharomyces, Candida, Schizosaccharomyces,
and Neurospora are fungi
Dictyostelium discoideum, Entamoeba,
Plasmodium falciparum, Trypanosoma, Giardia are protists
or protozoa
Sulfolobus acidocaldarius (70oC),Archaeoglobus
fulgidus (83oC), Methanosarcina barkeri (30-37 oC), Methanosarcina
mazeii (37oC), Methanococcus jannaschii (80oC), Haloferax
volcanii (37oC), Halobacterium salinarium (ca37oC), Methanobacterium
thermoautotrophicum (60-65oC), Desulfurococcus sp. (85-90 oC), Thermococcus
sp (75+ oC) (archaea),
Enterococcus hirae (37oC), Borrelia
burgdorferi (33-37oC), Thermus thermophilus (70-80oC), Deinococcus
radiodurans (30oC) (Bacteria) are prokaryotes. (Usually Bacteria have an
F- and not an A-ATPase. The bacteria probably obtained the archaeal/vacuolar type
ATPase through horizontal gene transfer.)
The
prokaryotes can be considered as outgroup for the eukaryotes.
Design a question that you can address using ml mapping.
For example:
Are the plants, green algae and red algae a monophyletic group?
Is Giardia the deepest branch among the eukaryotic sequences?
Do the animals group with the fungi, or do the fungi go with the plants?
After you have a question, figure out what your 4 groups should be (see the tree below). Only then start the program.Before entering the final y to run the program, select that you want to run this on all possible quartets (option n, enter 0) with one sequence from each group.
Select a gamma distribution with 8 classes, enter the shape parameter as 0.6.This is an exercise in thinking and concentration. If you think too little, you will have to go back to the start several times.
The eukaryotic part of the tree calculated with phyml is as follows, the whole tree is here:
6) Puzzleboot (only if you have time, and you want to try this).
puzzleboot is a UNIX shell-script program that allows the distance matrix option of PUZZLE to be used in the context of a bootstrap analysis with PHYLIP programs (something which PUZZLE was not originally designed to do).
Use a file of your choice (needs to be phylip formatted).
Copy puzzleboot_mod.sh
and puzzle.cmds into
the directory where you want to run the script. .
CAREFUL: puzzleboot removes
all outfiles and outtrees from the directory it runs in!
change permission
of puzzleboot_mod.sh
chmod u+x puzzleboot_mod.sh
Change the responses in puzzle.cmds to that they correspond to the model you want to use. You probably want to fix pinvar and alpha to values you already estimated!
Hint: to help trouble shooting, run the puzzleboot first on the original data (one file), move to the bootstrap sample only after you are happy with the commands file.
Run seqboot on your data. To execute the script type:
./puzzleboot_mod.sh
your_file_name_that_contains_the_bootstrapped_samples
The output consists of 100 distance matrices, run them through neighbor or fitch. You need to select the m option.
You get the support values with consense.