Dotlet
|



Go over goals from class 14
Comments on a "natural taxonomy".
Shared derived vs shared primitive characters, groups from homoplasies (see cladistics -- Ashlock (Ernst Mayr, Lynn Margulis, and others) versus Hennig (Woese, Pace, and others --- for discussion see here, here and here)
Here is exampe providing an impression on the heatedness of the ongoing depate: "Oddly, the school of ‘phylogenetic systematics’ founded by Hennig (1966) grossly downplayed the phylogenetic importance of progressive change compared with splitting, seen by them as so all-important that many Hennigian devotees dogmatically insist that ancestral groups like Bacteria, Protozoa and Reptilia be banned. Hennig called such basal groups with a monophyletic origin ‘paraphyletic’ and redefined monophyly to exclude them and embrace only clades, likewise redefined as including all descendants of their last common ancestor. This redefinition of ‘clade’ is universally accepted, but Hennig's extremely confusing and unwise redefinition of monophyly is not. Though accepted by many, sadly probably the majority (especially the most vociferous and over self-confident, and those fearful of bullying anonymous referees, of whom I have encountered dozens mistakenly insisting without reasoned arguments that paraphyletic taxa are never permissible), it is rightly firmly rejected by evolutionary systematists who consider the classical distinction between polyphyly and paraphyly much more important than distinguishing two forms of monophyly (paraphyly and holophyly, using the precise terminology of Ashlock (1971), where holophyletic equals monophyletic sensu Hennig)." from Tom Cavalier Smith http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2842702/
Introns and Their EvolutionThree groups of introns based on their splicing mechanisms:group I and II are self-splicing [have different splicing mechanism: see this figure for comparison of splicing]: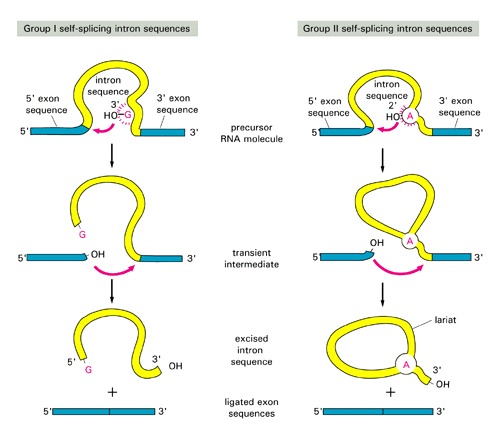 group III introns are present in eukaryotic nucleus, need spliceosomes to splice out: 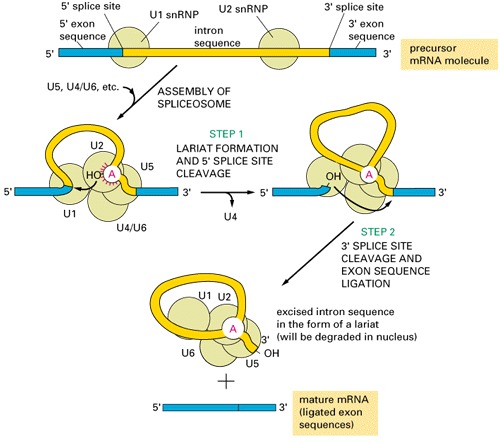 Where different groups of introns occur?
Where do spliceosomal introns come from and how the splicing machinery evolved?Hypothesis:Spliceosomal introns evolved from Class II introns; the function of some of the internal loops of the class II introns are taken over by the spliceosomal snRNA (small nuclear RNA).Support:
Gratuitous complexity hypothesis for evolution of spliceosomal machinery: See reading assignment on WebCT [the portions for the reading are highlighted in the PDF file] Problem:class II introns are found in bacteria, and only in one Archaeal genus, Methanosarcina; why is it that predominately "crown-group" eukaryotes have introns?Not much of a splice site consensus (exon1 GT-intron-AT exon2) Group I introns often have homing endonucleases. Also: reverse splicing Possible benefits of having introns:Exon shuffling, alternative splicing (1 gene -> different protein products) ....Two rival hypotheses: Intron Early vs. Intron LateIntron early:Protein diversity arose in analogy to exon shuffling in the generation of antibody diversity (see your biochemistry or genetics textbook on the maturation of the immune system).Claims:
Intron late:Present day introns are late invaders of already functional genes. Exon shuffling might play some role in eukaryotes, but most of protein diversity arose before introns invaded protein coding genes.Claims:
Compromise:mixed model of intron evolution
Else:it was suggested that class II introns were the reason for the separation between transcription and translation in Eukaryotes (accomplished through the nuclear envelope). Martin and Koonin's hypothesis suggests that class 2 introns were brought into the eukaryotic cell by the mitochondrial endosymbiont.
|
{kind=link}