Introns and Their Evolution
Three groups of introns based on their splicing mechanisms:
group I and II are self-splicing [have different splicing mechanism: see this figure for comparison of splicing]: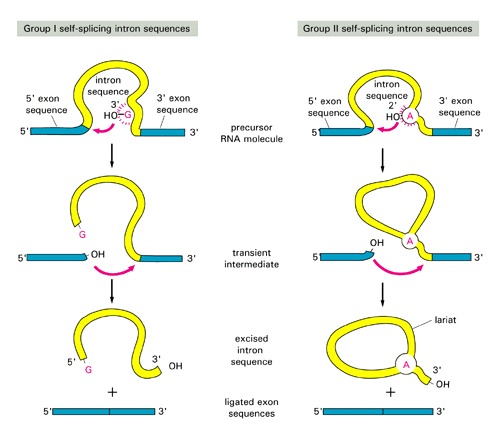
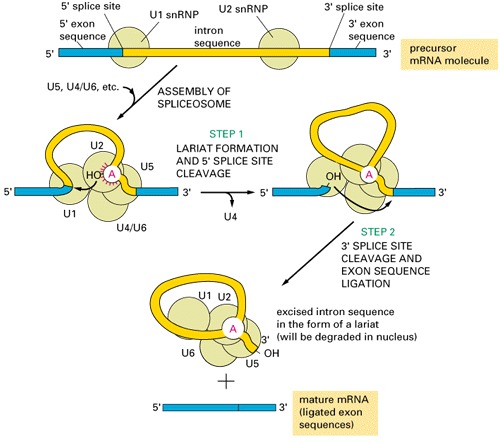
group III introns are present in eukaryotic nucleus, need spliceosomes to splice out:
Where different groups of introns occur?
- Group I: were discovered in ciliated protozoan Tetrahymena; found also in Physarum, fungal and algal and plant mitochondria and in phage T4, rare in Bacteria, one is present in Thermotoga 23SrRNA. Similar to inteins, they often rely on Homing Endonucleases to invade a host gene.
- Group II: common in Bacteria, and so far found only in one Archaeal genus, Methanosarcina
- Spliceosomal Introns: present throughout eukaryotes, but more common in "crown-group" eukaryotes
Where do spliceosomal introns come from and how the splicing machinery evolved?
Hypothesis:
Spliceosomal introns evolved from Class II introns; the function of some of the internal loops of the class II introns are taken over by the spliceosomal snRNA (small nuclear RNA).Support:
- Group II introns are often located in intergenic regions in Bacteria, suggesting their mobility as parasitic genetic elements
- Group II and spliceosomal introns both form a lariat structure (see figures above)
- class II introns that are non-functioning, because a loop has been removed, splice in the presence of snRNA.
- The reverse is true too: domain of a group II intron can substitute snRNA of the spliceosome
Gratuitous complexity hypothesis for evolution of spliceosomal machinery: See reading assignment on WebCT [the portions for the reading are highlighted in the PDF file]
Problem:
class II introns are found in bacteria, and only in one Archaeal genus, Methanosarcina; why is it that predominately "crown-group" eukaryotes have introns?Not much of a splice site consensus (exon1 GT-intron-AT exon2, see here for the splice site consensus in Arabidopsis)
{kind=link}
Group I introns often have homing endonucleases.
Homing endonucleases and intron mobility. Spread in populations, selective pressure on endonuclease. See the excellent paper by Goddard and Burt on the reinvasion cycle.
Also: reverse splicing
Possible benefits of having introns:
Exon shuffling, alternative splicing (1 gene -> different protein products) ....Two rival hypotheses: Intron Early vs. Intron Late
Intron early:
Protein diversity arose in analogy to exon shuffling in the generation of antibody diversity (see your biochemistry or genetics textbook on the maturation of the immune system).Claims:
- Introns separate structural domains. Example of a Go-plot is here (from here, these authors describe an significant excess of introns in the linker regions defined through he overlap in the Go-plot).
- In Triose Phosphate Isomerase an intron was found in a position suggested by a Go-plot (here).
- Introns arose early, before the uptake of the mitochondrial and chloroplast endosymbiont,
- Neighboring introns often are in the same phase. While significant, the excess is rather small: 216 of 570, 36 more than expected under a random distribution). However, the excess is larger, if only multidomain proteins are considered, suggesting that these indeed evolved through exon shuffling (see here for a recent analysis).
{kind=link}
Intron late:
Present day introns are late invaders of already functional genes. Exon shuffling might play some role in eukaryotes, but most of protein diversity arose before introns invaded protein coding genes.Claims:
- distribution of introns mapped on phylogenetic trees unambiguously points towards late invasion (and here).
- The correlation between structure and intron position is not unambiguous.
- The finding that introns in mitochondrial (eubacterial) and nucleocytoplasmic genes have introns in the same location could reflect a preferred intron integration site. The phase pattern is also observed in vertebrate genes, in which the introns are of late origin.
- Exon shuffling requires introns located in the same phase, but there might be other reasons for having a slight excess of introns in the same phase. For introns to frequently invade genes, there needs to be mechanisms for introns to find new "homes" (see above).
Compromise:
mixed model of intron evolution- version 1 - while some introns are recent, most are old. E.g.: [Roy, 2003].
- version 2 - while most introns are recent, some are older, but not necessarily very old. E.g.: [Rogozin et al., 2003]
Else:
it was suggested that class II introns were the reason for the separation between transcription and translation in Eukaryotes (accomplished through the nuclear envelope). Martin and Koonin's hypothesis suggests that class 2 introns were brought into the eukaryotic cell by the mitochondrial endosymbiont.