Optional exercises:
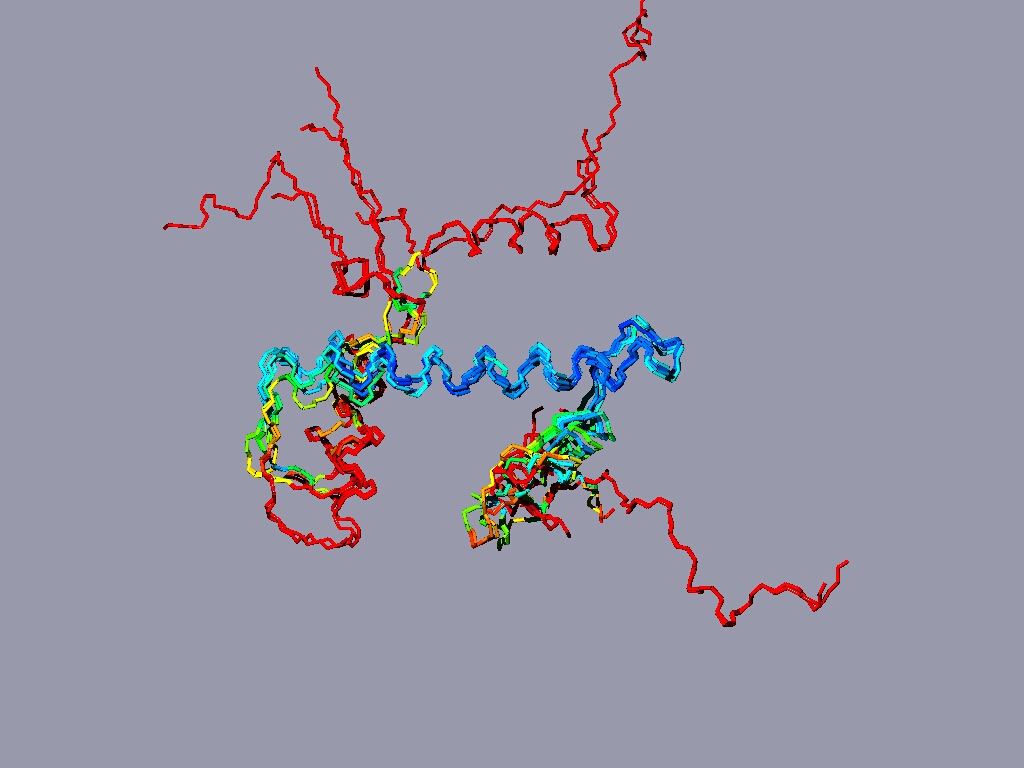
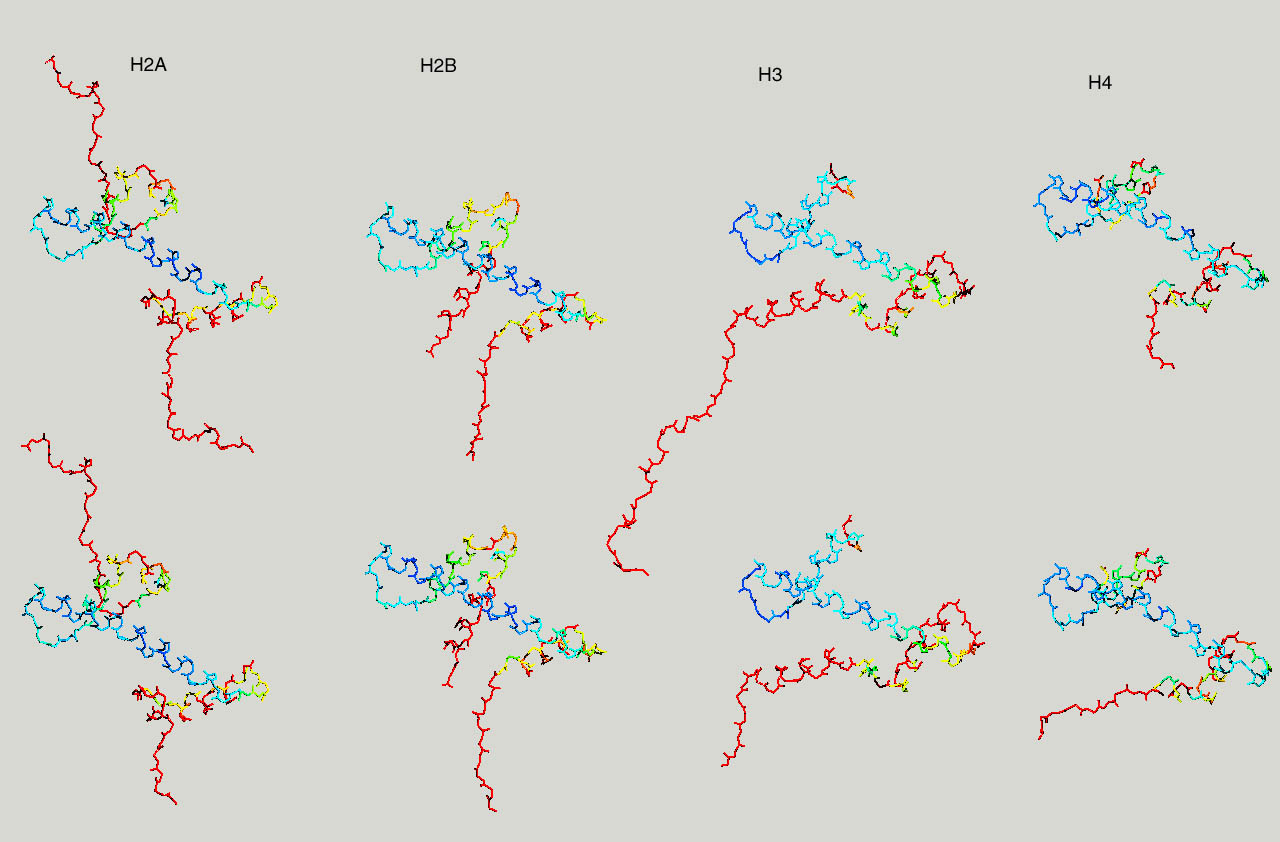
If you have more time to spare and you are up for a challenge, take a look at the nucleosome (1AOI.pdb). Open it from within chimera. Save each of the 8 histones as a separate pdb file. Close the nucleosome file, open the 8 histone files and align them to one reference histone.
Below same as last figure, but histones are depicted side by side :
Proteasomes are the recycling cans of the cell. Ubiquitin tagged protein are taken-up into the proteasome and broken down into aa. The proteasome (20S) core consists out of homologous alpha and beta subunits, arranged in 4 rings. The two central ones are beta subunits (2x7), the two rings at the ends are made from alpha subunits. Humans (and other eukaryotes) have 7 different genes encoding seven different alpha subunits, and 7 different genes encoding beta subunits, and each is present in duplicate in the 20S proteasome core. Altogether 28 subunits.
We are interested in creating a structure based sequence alignment of the different alpha and beta subunits. To do so, pick the bottom or top half of the proteasome (5LF7.pdb, or a similar high resolution structure from another organism), and save all the different subunits as separate pdb files (if you use 5LF7.pdb, the pdbs for the individual SU are here). First align all the alpha beta subunits separately, and then try to generate an alignment of the alpha with the beta subunits. Use matchmaker to align the structures, then use Tools > structure comarison > Match-Align to create a multiple sequence alignment.
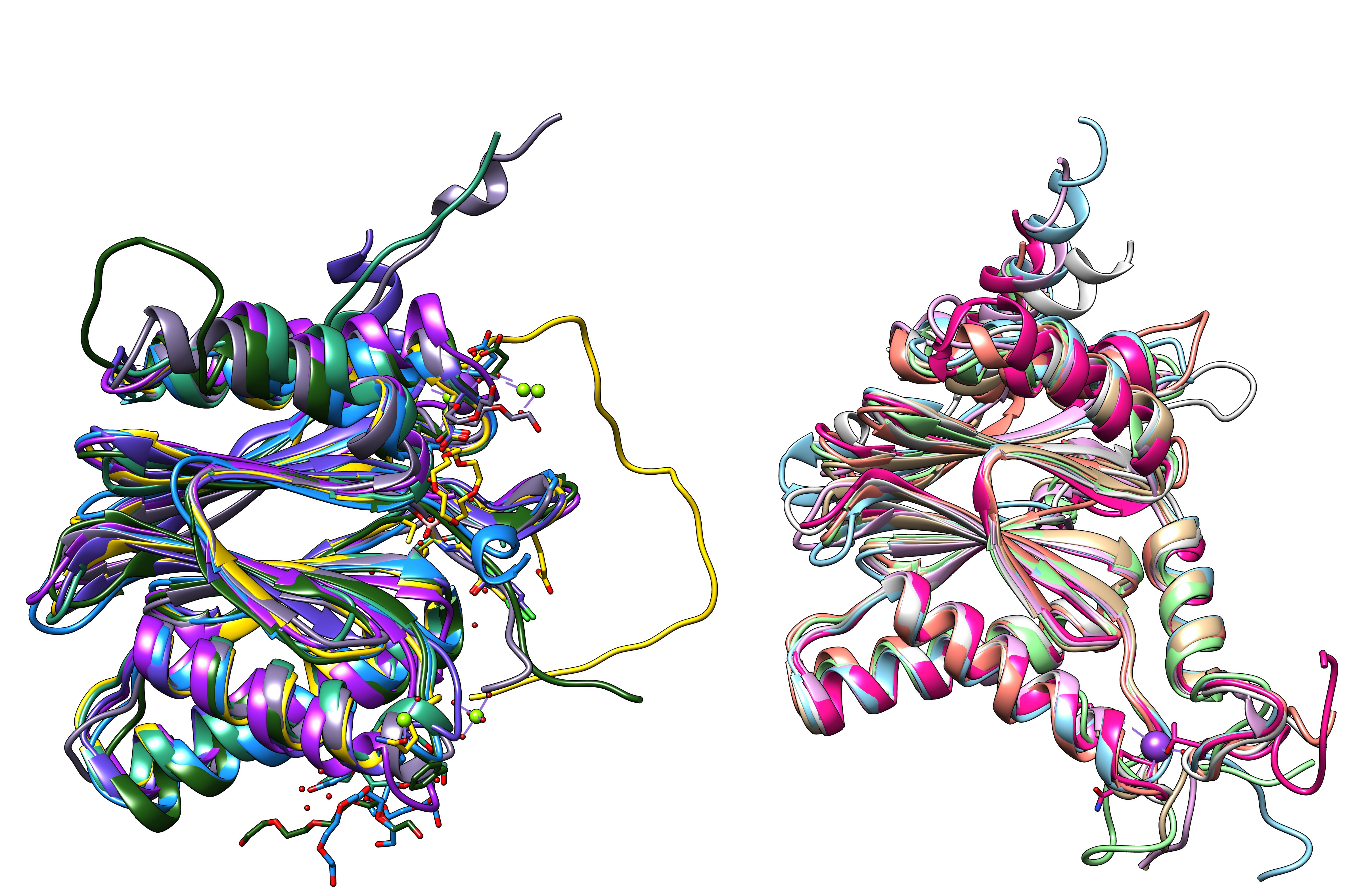
Proteasome subunits (7 beta and 7 alpha subunits) from human hela cells (5LF7.pdb)

All 14 subunits of the human proteasome core superimposed.