MCB 5472 : MrBayes and dN/dS
1) Introduction
to Bayesian Analyses
We will use MrBayes3.2 as installed on the bioinformatics cluster. To start the program, move to the directory where your sequence data are located and type mb.
Skip this paragraph: The latest version of the MrBayes software for different operating systems and a link to the MrBayes manual is available here.
Exercise
1: Goal of this exercise is to
learn how to use MrBayes to reconstruct phylogenies.
If you already are familiar with MrBayes, skip this exercise!
- Save the file ATPasSU_2013.nxs into a folder on the cluster. The dataset is of ATPase subunits you used earlier, but in
the NEXUS format that MrBayes reads (the nexus format is used by PAUP and many other programs - it allows to pass trees and commands to the different programs -- clustal, seaview, and many other programs write nexus formated files, but usually, you need to go into the file with a text editor and change things like the way gaps are treated, or the data type).
For more info on the dataset see
http://carrot.mcb.uconn.edu/mcb3421_2013/assign10.html. You might need to rename the file after downloading. Have a look at the file in a text editor and make sure you know the name of the file and the directory where the file is stored (often difficulties arise because one program or the other has added an extension to the file). Start a terminal connection to the bioinformatics cluster, execute the qlogin command to be assigned to a compute node, change the directory to the directory where you stored the nexus files, and start MrBayes through typing mb at the command-line.
- At the MrBayes
command prompt type "execute ATPaseSU_2013.nxs". This will load the data into the
program.
- Here is an explanation of the commands at the end of sequence file in the MrBayes block:
prset aamodelpr=fixed(jones) [this sets the substititution matrix to JTT, a mdern version of PAM]
lset rates=gamma [this selects the gamma distribution to describe ASRV - this makes the program run a lot slower, you might not want to execute this option]
mcmcp samplefreq=100 printfreq=100 [this sets the frequency with which the "robot" reports results to the screen and to the files (different files for parameters (.p) and trees (.t))]
mcmcp savebrlens=yes [mcmcp sets parameters for the chain. savebrlens tells it to save the trees with branchlengths]
[mcmcp filename=ATPaseSU.nexus] [if you use this command, it tells the program to save the files under a certain name. This is handy, if you want to read in data from a previous run, but it usually is easier to go with the default]
mcmcp ngen=5000 [this sets the number of generations to run the chain for to 5000 generations]
" prset" sets the priors for the model used to calculate likelihoods. In this case we choose the substitution parameters from the JTT amino acid substitution model (Jones et al., 1992).
" mcmcp " sets parameters
for Markov Chain Monte Carlo: we set to sample every 50 generation, to print results
to a screen every 50th generation, run 2 chains simultaneously, start with random
tree, and save branch lengths.
- Type "showmodel" to have MrBayes display the model you selected.
- At the MrBayes command prompt type "mcmc". This actually runs the chains, and we set it to run for 5000 generations.
The program runs two analyses in parallel (by default each with 4 chains, and three of these chains are heated; it definitely is a good idea to run mb on a fast and remote computer). The smaller the reported average standard deviation of split frequencies is, the more reliable the result (i.e., your run is close enough to infinitely long). When the value is below .015, or when your patience is exhausted, terminate the run, by typing no at the prompt.
This might be a good point in time to check the MrBayes manual
- Make sure you have typed "no" at the Continue with analysis? (yes/no): prompt.
- After the run is finished, the " sump " command will
plot the logL vs. generation number, that allows to determine the necessary burnin (you want to discard those samples as "burnin" where the -logL is still rising steadily).
[Rather than using the sump command, you also can import the parameter file into EXEL and plot the logL values as a chart in EXEL. See below.]
At the start of the run, the likelihood rapidly increases by orders of magnitude. If the first samples are included in the plot, one really cannot see if the likelihood values fluctuate around a constant
value. You can exclude the first couple of samples by specifying a burnin. (The new version of MrBayes uses a burnin of 25% by default.)
sump relburnin=no burnin=20 excludes the first 20 samples (as we sampled only every 100 trees, a burnin of 20 excludes the first 2000 trees).
Type
sumt relburnin=no burnin=20
, where you need to substitute
'20' with the number you obtained in the previous step of the exercise. This command creates a consensus tree, and shows posterior
probabilities of the clades. You can take a look at the tree on the screen, or in figtree, or njplot by
loading the ATPaseSU_2013.nxs.con file into these programs.
Which value did you use for the burnin?
Which branch in the tree is the longest? (check the bipartition and branch lengths tables) How long is it?
What is the measure?
Are there features of the tree that you find surprising? If yes, what does this tell you about the posterior probabilities calculated by MrBayes?
Can
you explain in a few words, why is it important to exclude a 'burnin' from our analyses?
Type " quit " at the prompt to exit MrBayes.
If you use MrBayes in your research, you want to sample as many generations in as many parallele runs as possible. You can increase the computational power, by assigning different chains to different nodes. This can be done by running the mpi version of mrbayes. A shell script that submits an mb-mpi job to the queue is here. More instructions on the queue are here and here
2. MrBayes
by example: Identification of sites under positive selection in a protein
Background:
Professor Walter M. Fitch and assistant research biologist Robin M. Bush
of UCI's Department of Ecology and Evolutionary Biology, working with researchers
at the Centers for Disease Control and Prevention, studied the evolution of a
prevalent form of the influenza A virus during an 11-year period from 1986 to
1997. They discovered that viruses having mutations in certain parts of an important
viral surface protein were more likely than other strains to spawn future influenza
lineages. Human susceptibility to infection depends on immunity gained during
past bouts of influenza; thus, new viral mutations are required for new epidemics
to occur. Knowing which currently circulating mutant strains are more likely to
have successful offspring potentially may help in vaccine strain selection. The
researchers' findings appear in the Dec. 3 issue of Science magazine.
Fitch
and his fellow researchers followed the evolutionary pattern of the influenza
virus, one that involves a never-ending battle between the virus and its host.
The human body fights the invading virus by making antibodies against it. The
antibodies recognize the shape of proteins on the viral surface. Previous infections
only prepare the body to fight viruses with recognizable shapes. Thus, only those
viruses that have undergone mutations that change their shape can cause disease.
Over time, new strains of the virus continually emerge, spread and produce offspring
lineages that undergo further mutations. This process is called antigenic drift.
"The cycle goes on and on-new antibodies, new mutants," Fitch said.
The
research into the virus' genetic data focused on the evolution of the hemagglutinin
gene-the gene that codes for the major influenza surface protein. Fitch and fellow
researchers constructed "family trees" for viral strains from 11 consecutive flu
seasons. Each branch on the tree represents a new mutant strain of the virus.
They found that the viral strains undergoing the greatest number of amino acid
changes in specified positions of the hemagglutinin gene were most closely related
to future influenza lineages in nine of the 11 flu seasons tested.
By studying
the family trees of various flu strains, Fitch said, researchers can attempt to
predict the evolution of an influenza virus and thus potentially aid in the development
of more effective influenza vaccines.
The research team is currently expanding
its work to include all three groups of circulating influenza viruses, hoping
that contrasting their evolutionary strategies may lend more insight into the
evolution of influenza.
Along with Fitch and Bush, Catherine A. Bender,
Kanta Subbarao and Nancy J. Cox of the Centers for Disease Control and Prevention
participated in the study.
A talk by Walter Fitch (slides and sound) is here
Exercise:
The goal of this exercise
is to detect sites in hemmagglutinin that are under positive selection.
Since
the analysis takes a very long time to run (several days), here are the saved
results of the MrBayes run: Fitch_HA.nex.p , Fitch_HA.nex.t .
The original data file is flu_data.paup . The dataset is obtained from an
article by Yang et al, 2000.
The MrBayes block used to obtain results above is:
begin
mrbayes;
set autoclose=yes;
lset nst=2 rates=gamma nucmodel=codon omegavar=Ny98;
mcmcp samplefreq=500 printfreq=500;
mcmc ngen=500000;
sump burnin=50;
sumt burnin=50; end;
Selecting a nucmodel=codon with Omegavar=Ny98 specifies a model in which for
every codon the ratio of the rate of non-synonymous to synonymous substitutions
is considered. This ratio is called OMEGA. The Ny98 model considers three different
omegas, one equal to 1 (no selection, this site is neutral); the second with omega
< 1, these sites are under purifying selection; and the third with Omega >1,
i.e. these sites are under positive or diversifying selection. (The problem of
this model is that the there are only three distinct omegas estimated, and for
each site the probability to fall into one of these three classes. If the omega>1
is estimated to be very large, because one site has a large omega, the other sites
might not have a high probability to have the same omega, even though they might
also be under positive selection. This leads to the site with largest omega to
be identified with confidence, the others have more moderate probabilities to
be under positive selection).
Note : Version 2.0 of Mr Bayes has a model that estimates omega for each site individually,
the new version only allows the Ny98 model as described above..
- First,
you need to detect how many generations to burn in (meaning the number of samples you will have to discard). Import the file Fitch_HA.nex.p.txt into
Excel (link is above, open a new spreadsheet, select DATA, get external data, import text file (if your file does not have a txt extension, select enable all document types) -- select windows format, else defaults; (if you use an older version of Excel you need to import the file into two separate worksheets - it has too many columns; to import the remaining columns of the spreadsheet see 2. below) and plot # of generations versus -LnL values. Determine after how many generations
the graph becomes "stationary". The burnin value is that number of generations divided
by 50 (since only every 50th generation was sampled; i.e. the burnin value roughly
is equal to the number of rows - not quite because there is a header). To more
accurately determine the burnin, you need to rescale the Y-axis (click at the Y-axis -- if you aim accurately, you'll get a box that allows rescaling).
The result (scatterplot of LogL versus generation) might look like this:
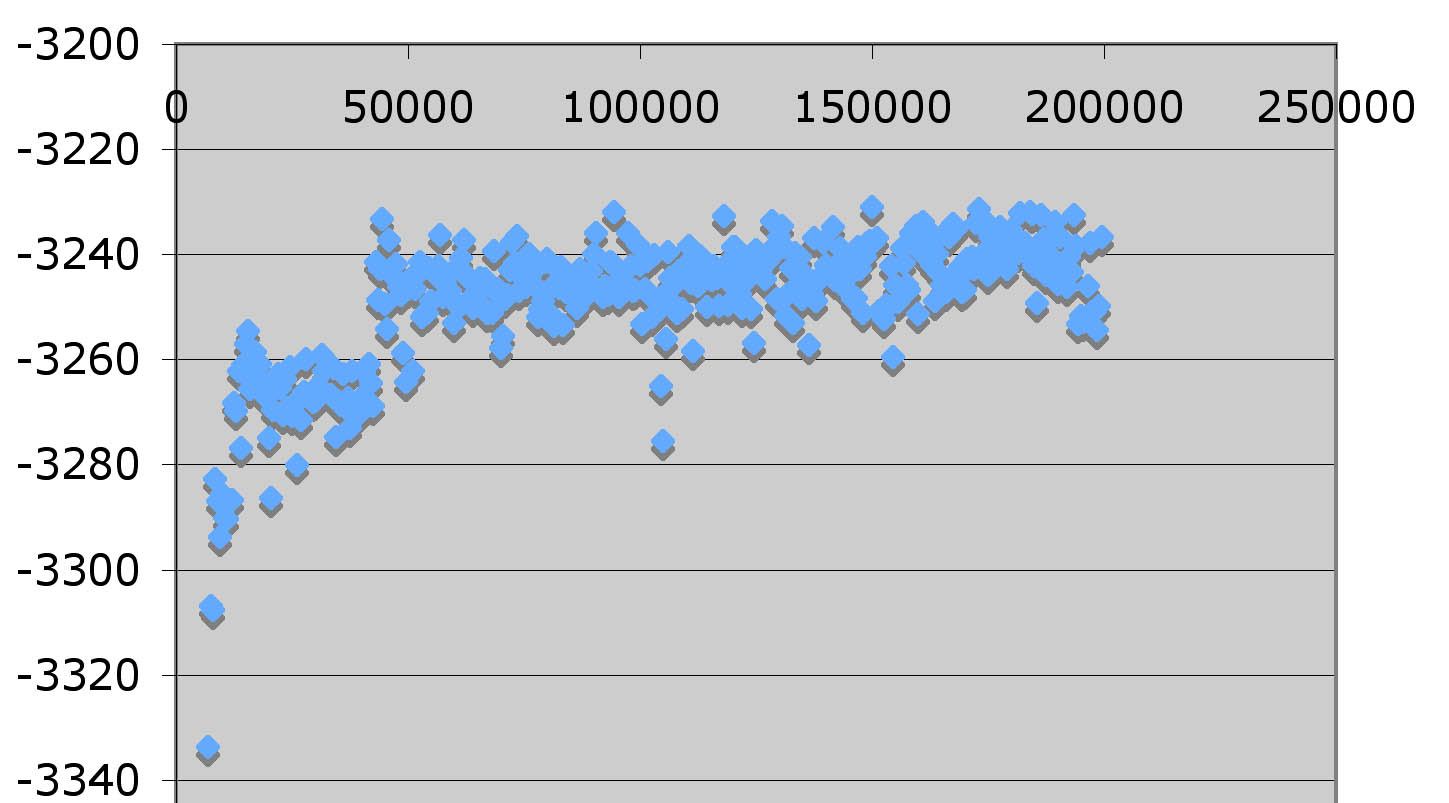
- Load Fitch_HA.nex.p into Excel. Go to Data->Get External Data->Import Text File. (In older versions of Excel, you need to load the data into 2 sheets, sincethe old Excel does not allow
to load more than 256 columns per sheet. To load the data, create a new Excel
spreadsheet. Go to Data->Get External Data->Import Text File , and load
first 256 columns.
Go to a separate sheet, and repeat "Get External Data"
command to load the rest of the data, you need to block out (=select =make black)
and exclude the first 256 columns (the last imported codon ended on nuc 555) -- you need to click the radio button AFTER you selected the columns to skip!). This file contains information for posterior
probabilities for each codon (columns) at each sampled generation (rows).)
- Calculate average posterior probability for each site of being under positive
selection (Do not forget to exclude first N rows as a burnin; you should have
detected value of N in the first question of this exercise - to be clear on where the burnin ends, you might want to highlight the rows representing the burnin and select a different font color. (Use AVERAGE() function
of Excel, enter the formula in a cell below the values for the individual trees -- starting in column pr+(1,2,3) --
copy the formula to all columns)
- Plot average posterior probability
vs. site # . (select the row in which you calculated the averages, then click
Graph, and select a bar graph). Write down the codon positions for a few sites
with the highest posterior probability of being positively selected (the columns
name pr+(1,2,3), pr+(4,5,6)....and so on. pr+(1,2,3) mean probability of codon
#1 (nucleotide #1, #2 and #3) to be under positive selection))
- Determine the 90% credibility interval
for the omega<1 value. To do this you sort posterior probability column in
ascending order (Select data you want to sort, and go to Data->Sort... ). Again,
do not forget to discard the burnin ; the easiest might be to actually delete
it.. After sorting, exclude 5% of the data on the top and on the bottom. The
range of the remaining data gives you the 90% confidence interval. (Enter answer in box below!)
Which codon was inferred to have a high posterior probability to be under diversifying selection?
What is the
90% credibility interval for the omega for sites under purifying selection?
3) dN/dS using codeml
You can use a dataset of you choice for this exercise (you will need a codon based alignment in phylip format -- non-interleaved, sequence designations 10 characters - if interleaved, add an I to the 1st line) , or you can use these files:
hv1.phy
hv1.tree
codeml.hv1.sites.ctl
codeml.hv1.branches.ctl
(the hv1 data are neurotoxins produced by spiders, see here for more information)
or
codeml.Bsub.ctl
Bsub_holin_del_codeml_tree.nwk
Bsub_holin_aln_del_codeml.phy (note the I in the header of the file)
The branch model for the hv1 dataset takes way too long to run in class.
1) decide which analyses you want to perform, and modify the control file accordingly, run the analysis (> codeml codeml.XYZ.ctl). If in doubt, check the PAML manual at http://abacus.gene.ucl.ac.uk/software/paml.html.
2) if you have time, try to analyze the same dataset at the datamonkey. (click on analyze your data in the header, when done click the links to the different output files.)
Which dataset did you analyze?
How many codons were inferred to be under purifying selection?
Using the LRT, is the more complicated model justified?
4) Work on your student project!
Include a one sentence summary of what you did in your email.